Lit.: F. Cvrčková: Praktický úvod do bioinformatiky, Akademia, 2007.
A B C D E F G H I J K L M N O P Q R S T U V W X Y Z
|
A top Ab initio Ab initio Výpočet bez použití experimentálních dat, pouze na základě teorie. 1. Predikce struktur proteinů bez templátu. 2. Soubor kvantově-chemických metod. (1. a 2. nemají nic moc společného) Anfinsenova hypotéza Anfinsen's dogma Tato teorie předpokládá, že nativní konformace proteinů (alespoň malých) je zakódována v aminokyselinové sekvenci a to tak, že představuje minimum volné Gibbsovy energie. Nativní konformace je tedy unikátní, stabilní a kineticky dostupná. Existují proteiny, kde se zdá, že tato teorie neplatí, že aktivní konformace je metastabilní (např. inhibitory proteas serpiny). Anotace Annotation Identifikace genu na základě podobnosti sekvencí nebo dalších parametrů. Může být i experimentální. B top Binární přiřazení sekvencí Binary sequence alignment Vzájemné přiřazení dvou sekvencí. Bioinformatika Bioinformatics Bioinformatika se zabývá např. zpracováním dat sekvenačních projektů, anotací genomů, studiem evoluce, analysou exprese, předpovědmi prostorových struktur proteinů, studiem biomolekulárních interakcí a podobně. Na bioinformatiku navazují např. systémová biologie a chemoinformatika. BLAST BLAST (Basic Local Alignment Search Tool) Základní program pro prohledáváni sekvenčních databází pomocí sekvenčního dotazu. Existují různé varianty podle typu dotazu. Dále existují vylepšené iterativní verze. ? Bootstraping Hodnocení algoritmů tak, že se mu předhodí nějakým způsobem randomizovaná data. Používá se např. při studiu fylogenetických vztahů. C top CAPRI CAPRI Projekt podobný CASP, ale zaměřený na predikce interakcí protein-protein. CASP CASP Projekt hodnotící metody predikce prostorových struktur proteinů. Nejprve jsou vybrány nějaké zajímavé proteiny, jejichž prostorová struktura není známa. Struktury potom paralelně měří experimentátoři a předpovídají teoretici. Teoretici musí své predikce odevzdat dříve, než experimentátoři. Ti, kteří dokáží struktury nejlépe predikovat, získají mezinárodní uznání. Cena mezer Gap penalty Jeden ze vstupních parametrů pro programy pro konstrukci přiřazení sekvencí. Nastavení ovlivňuje počet a velikost mezer v přiřazení. Rozlišujeme gap open a gap extension penalty. CLUSTAL CLUSTAL Program pro konstrukci mnohočetného sekvenčního přiřazení. Cluster Klastr Shluk. 1. Při shlukové analýze dat. 2. Paralelní počítač sestavený z komoditního hardwaru. ? Coarse graining Přístup, kdy se při simulacích systém nepopisuje prostřednictvím jednotlivých atomů, ale prostřednictvím částic, kde každá částice představuje skupinu atomů. Umožňuje simulovat delší časy a/nebo větší systémy. 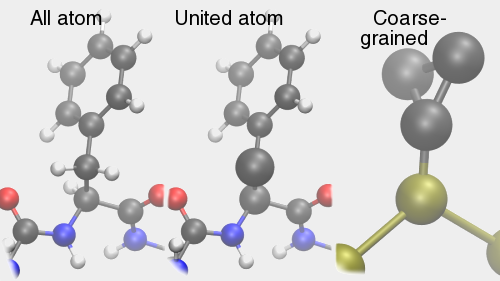 Connollyho plocha Connolly surface Též rozpouštědlem vyloučená plocha. Plocha získaná tak, že kolem molekuly "obtáčíme" sondu, představující rozpouštědlo. Connollyho plocha je pak definovaná tím, kam až se sonda dostane. ? Constraint Strukturní omezení při molekulární simulaci nebo minimalizaci energie. Obdoba restrainu, akorát na pevno. D top DDBJ DDBJ Jedna z hlavních sekvenčních databází. De novo De novo Viz Ab initio. Dekonvoluce Deconvolution Opak konvoluce. Pojem konvoluce/dekonvoluce se používá v mnoha významech. Například pokud smícháme několik sloučenin dohromady a pak testujeme biologickou aktivitu směsi, jedná se o konvoluci. Při dekonvoluci zjišťujeme která ze sloučenin ve směsi je aktivní. Matematicky je konvoluce dvou funkcí f(x) a g(x) definována jako integrál přes celý prostor součinu f(x) a g(x-t) a je to funkce proměnné t. Uplatnění má v NMR (Fourierova transformace součinu funkcí je konvolucí jejich Fourierových transformací). U mikroskopických technik je konvolucí kombinace reálného obrazu a optické chyby. Pokud si u mikroskopu nakalibrujeme optickou chybu, je pak možné dekonvolucí získat přesnější obraz. Dendrogram Dendrogram Fylogenetický strom. Grafické znázornění příbuzenských vztahů mezi sekvencemi. Distribuované výpočty Distributed computing Provedení výpočtu jeho rozložením na několik počítačů. Na rozdíl od clusterů a superpočítačových center se jedná o heterogenní prostředí, např. kancelářské počítače ve farmaceutických firmách nebo počítačích dobrovolníků (např. Folding@home). Jaký je rozdíl mezi distribuovanými výpočty a gridy není nejspíš nikomu známo. ? Docking 1. Předpověď struktury nějakého komplexu, např. protein-protein nebo protein-ligand. Zajímá nás struktura komplexu a síla vazby, ale zpravidla nás nezajímá proces vazby. 2. Samotný proces vazby. Dolování dat Data mining Analýza rozsáhlých datových souborů s cílem je převést na použitelné hypotézy. Doména Domain Část proteinu která je strukturně oddělená (samostatná globule) ale je pořád součást í jednoho polypeptidového řetězce. Někdy může být doména definovavá funkčně. Dotaz Query Dotaz zadaný programu při prohledávání databáze. Může se jednat o heslo (např. pro sequence retrieval system) nebo sekvence (BLAST). DSSP DSSP Definition of secondary structure of proteins(?) Všeobecně přijatá definice sekundárních struktur (tedy program, který přiřadí jednotlivým aminokyselinám typ sekundární struktury na základě struktury prostorové). E top E-hodnota E-value Hodnota, které charakterizuje podobnost sekvence nalezené pomocí programu pro prohledávání databází (např. BLAST). Je definovaná jako pravděpodobnost, že by byla stejně podobná sekvence nalezena ve stejně veliké databázi náhodých sekvencí. Elektrostatický potenciál Electrostatic potential Potenciální energie, vypočtená pro jednotlivé body v prostoru v okolí molekuly. Elektrostatický potenciál v určitém bodě odpovídá energii nutné k tomu, abychom tam dostali bod s jednotkovým nábojem. EMBL EMBL European Molecular Biology Laboratory. 1. Organizace. 2. Jedna z hlavních sekvenčních databází. Empirický potenciál Empirical potential Viz Force field. Epigenetika Epigenetics Vše co se vymyká možnosti zápisu v nukleotidové sekvenci DNA. Nejčastěji se tak označuje např. methylace DNA, změny struktury chromatinu atd. Epigenomika Epigenomics Systematické studium epigenetiky (viz) daného organismu. Explicitní rozpouštědlo Explicit solvent O explicitním rozpouštědlu mluvíme, pokud simulovanou molekulu (solut) obalíme jednotlivými molekulami rozpouštědla. Opak implictního rozpouštědla. 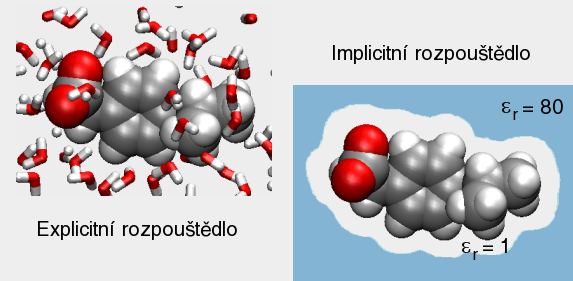 F top Farmakofor Pharmacophore Trojrozměrné uspořádání atomů (malé) molekuly, které je zodpovědné za její biologickou aktivitu (zpravidla vazbu na cíl léčiva). FASTA FASTA 1. Program sekvenčního prohledávání databází. 2. Způsob zápisu sekvencí. První řádek začíná znaménkem ">", který je následován označením sekvence. Na dalších řádcích se uvádí sekvence (nukleotidová, nebo aminokyselinová v jednopísmenných zkratkách). Je možné ho použít i pro několik sekvencí nebo pro přiřazení sekvencí. ? Fold Ukázalo se, že existují strukturně velmi podobné, ale sekvenčně velmi nepodobné skupiny proteinů. Jinými slovy, rozmanitost prostorových struktur je daleko menší než je rozmanitost aminokyselinových sekvencí a existuje jen relativně nízký počet typů prostorových struktur - "foldů". ? Fold recognition Pokud námi studovaný protein nemá triviálně detekovatelnou podobnost s proteinem, jehož prostorová struktura je známa, je možné ho vhodným postupem přiřadit k nějakému "foldu" (viz) a jeho strukturu předpovědět podle nějakého proteinu daného foldu se známou prostorovou strukturou. Jinými slovy se jedná o hledání homologie tam, kde žádná neni :-). ? Folding funnel Konformační plocha volné energie proteinu, tedy závislost Gibbsovy volné energie proteinu na konformaci. Obvykle se zobrazuje jako závislost volné energie na konformační entropii. Folding@home Folding@home Úspěšný grid (viz) projekt, zaměřený na simulaci sbalování proteinů. Funkční genomika Functional genomics Teoretické, ale častěji spíše experimentální studium biologických funkcí genů pomocí např. siRNA. Aby to byla -omika, musí to být systematické a vysoko-průtočné. Fylogenetický strom Phylogenetic tree Viz Dendrogram. G top GeneBank GeneBank Jedna z hlavních sekvenčních databází. Genetický algoritmus Genetic algorithm Původně byl vývoj těchto algoritmů motivován snahou modelovat evoluci, ale ukázalo se, že je možné je použít jako obecné optimalizační algoritmy. V souvislostí s touto metodou se používá populačně genetická terminologie (mutace, populace atd.) ? Grid Viz Distribuované výpočty. H top Harmonický potenciál Harmonic potential Potenciál pro popis kovalentních vazeb mezi dvěma atomy, případě valenčních úhlů. Potenciál závisí na vzdálenosti s tvarem k (r - r0)2, kde r0 je rovnovážná vzdálenost (případně úhel). 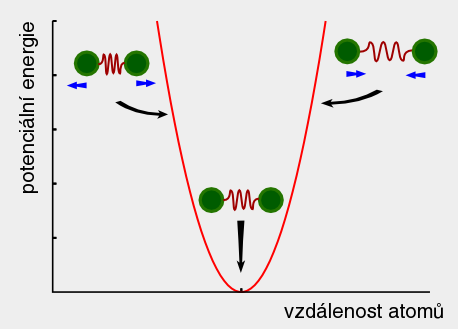 ? High Throuput Screening Paralelní testování mnoha potenciálních aktivních molekul, například potenciálních léčiv. O "vysokém průtoku" je možné mluvit od formátu mikrotitrační destičky, ale spíše se jedná o robotizované postupy, schopné testovat tisíce sloučenin v krátké době. Homologie Homology 1. Obecně podobnost sekvencí. 2. Kvantitativně označujeme homologii jako součet identity (dvě aminokyseliny pod sebou v přiřazení jsou identické) a podobnosti (dvě aminokyseliny jsou podobné - např. Asp a Glu). Homologní modelování Homology modelling Predikce struktury proteinu, která vychází z předpokladu, že proteiny s podobnou sekvencí mají podobnou prostorovou strukturu. I top Identita Identity Pro objasnění pojmů identita, podobnost a homologie, viz Homologie. Implicitní rozpouštědlo Implicit solvent O implicitním rozpouštědlu mluvíme, pokud simulovanou molekulu umístíme do kontinuálního prostředí, které napodobuje svými dielektrickými a hydromechanickými vlastnostmi reálné prostředí. Opak explictního rozpouštědla. In silico In silico Označení "na počítači". Často je kritizováno, že na rozdíl od pojmů in vitro, in vivo či in situ se nejedná o pojem latinský, ale preudo-latinský. Iterace Iteration Série kroků, kdy se nejprve vychází z odhadnutého výsledku a ten se v každém dalším kroku zpřesňuje. K top Kontig Contig Kontinuální sekvence získaná složením více výstupů ze sekvenátorů. Konvergence Convergence Konvergence nastává, pokud nějaký iterativně řešený problém spěje k jednomu výsledku. Při iterativních algoritmech se vychází z odhadu výsledku a v každém cyklu se přibližné řešení zpřesňuje. L top ? Lead compound Sloučenina nebo strukturní motiv, u něhož je experimentálně potvrzená žádaná biologická aktivita. Mezistupeň při vývoji léčiv. ? Lead optimization Proces vývoje léčiva, kdy je lead compound vylepšována tak, aby získala vlastnosti léčiva (vysoká afinita k cíli, nízká toxicita atd.). Lennard-Jonesův potenciál Lennard-Jones potential Potenciál pro popis nekovalentních van der Waalsovských (Dispersních) interakcí mezi dvěma atomy. Potenciál závisí na vzdálenosti s tvarem C12 r-12 - C6 r-6. 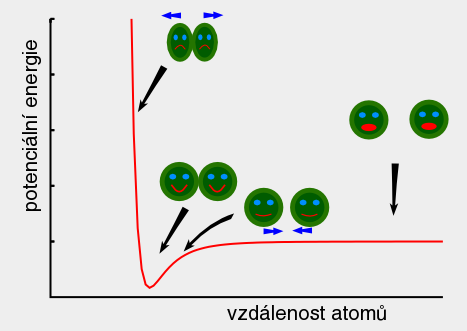 Levinthalův paradox Levinthal's paradox Myšlenkový postup Cyruse Levintala. Dokazuje, že proteiny nemohou hledat svou nativní strukturu systematickým prohledáváním všech možných struktur (pak by to trvalo miliardy let), ale i proces sbalování je zapsán v sekvenci. M top Mezery Gaps Místo v přiřazení sekvencí, které odpovídá deleci v genu, kde je mezera (respektive inzerci kde mezera není). Ze strukturního hlediska nejčastěji najdeme mezeru ve smyčkách na povrch proteinu, kdy v proteinu bez mezer je smyčka delší a s mezerou kratší. ? Missalignment Chybné sekvenční přiřazení. Mnohočetné přiřazení sekvencí Multiple sequence alignment Přiřazení více sekvencí. Model Model 1. Popis reality. 2. V souvislosti s predikcemi struktur proteinů se jako model označuje protein, jehož strukturu chceme předpovědět (narozdíl od templátu). Modelování Modelling (Engl.), Modeling (USA) Někdy je rozlišováno mezi modelováním a simulací tak, že v prvním případě se jedná o napodobování experimentu, kdežto v druhém případě se jde mimo podmínky experimentu. Například pokud změříme jak se budova hýbe ve větru a pak se budeme pokoušet spočítat to tak, aby to odpovídalo experimentu, jedná se modelování. Pokud v našem modelu nastavíme rychlost větru na uragán a budeme počítat kdy spadne, jedná se o simulaci. Modelování volné energie Free energy model(l)ing Metody, zaměřené na predikci hodnot změn volné (Gibbsovy, Helmholzovy) energie studovaných procesů. Moderovaná databáze Curated database Databáze, kterou spravuje člověk z masa a kostí. Molekulární plocha Molecular surface 1. Obecně nějaká plocha molekuly. 2. Viz Connollyho plocha. 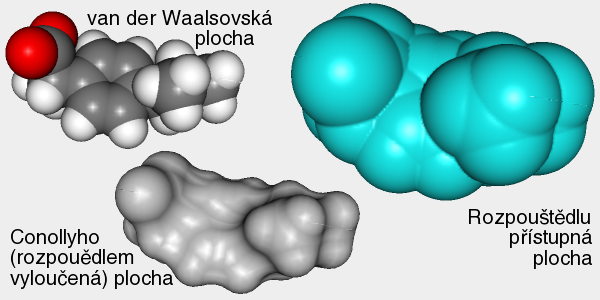 Monte Carlo Monte Carlo Algoritmy, které využívají počítačem generovaná náhodná čísla. Například hodnotu Ludolfova čísla můžeme vypočítat metodou Monte Carlo tak, že generujeme náhodná čísla x a y v rozsahy -1 až 1 a počítáme kolik párů x-y se trefí do kruhu s poloměrem 1 (x2 + y2 < 1). Podíl bodů uvnitř kruhu z celkového počtu bodů by se měl blížit hodnotě π/4. V bioinformatice a molekulárním modelováním se MC používa pro optimalizace a simulace molekulových systémů. Motiv Motif Úsek sekvence proteinu s nějakou biologickou funkcí, obvykle několika aminokyselin (kratší než doména). N top Navíjení Threading Metoda predikce struktury proteinu (v kategorii fold recognition). Aminokyselinová sekvence je "navíjena" na různé myšlené model struktur a je vybrán ten s nejlepším skóre, například pokud má realistické rozmístnění hydrofobních a hydrofilních aminokyselin. Needlemanův-Wunchův algoritmus Needleman-Wunch algorithm Nejstarší algoritmus pro konstrukci binárního přiřazení sekvencí. Poskytuje optimální přiřazení, ale je pomalý. Neuronová síť Neural network Původně se jednalo o snahu modelovat činnost nervové soustavy, ale později se začaly nervové sítě používat pro řešení různých problémů. Nervové sítě je možné "natrénovat" tak, aby dokázali rozpoznávat podobné objekty, jejichž podobnost není možné triviálně vyjádřit, například lidské tváře na fotografiích. V bioinformatice se používají např. při predikci sekundárních struktur. O top Optimalizace geometrie Geometry optimization Postup, při kterém se upravuje geometrie (souřadnice atomů) tak, aby měl systém co nejmenší potenciální energii. Většinou směřuje pouze do lokálního minima (optimalizací geometrie tudíž asi nejde dostat nativní struktury proteinu ze struktury rozbalené). Ortholog Ortholog Dva geny jsou orthology, pokud vznikly evolučním rozdělením druhu na dva druhy. Jinými slovy jsou to vzájemně sekvenčně homologní geny v genomech dvou druhů. Otevřený čtecí rámec Open reading frame Úsek DNA, který potenciálně kóduje protein. P top Paralog Paralog Dva geny jsou paralogy, pokud vznikly zdvojením genu v rámci evoluce daného organismu. Jinými slovy jsou to vzájemně sekvenčně homologní geny v jednom genomu. Parciální náboj Partial charge Náboje jednotlivých atomů molekuly. Neexistuje jednotný způsob jak je vypočítat, neboť existují různé definice parciálních nábojů, lišících se zjednodušeně řečeno v tom, jaká část elektronového obalu patří ke kterému atomu. 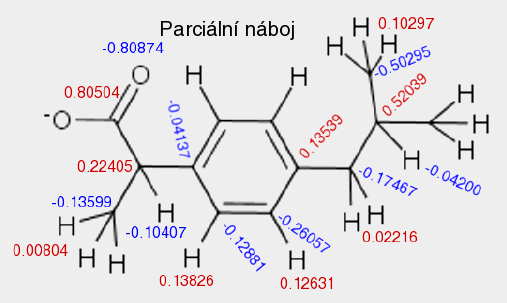 PDB PDB Protein databank. 1. Databáze prostorových struktur proteinů. 2. Formát zápisu prostorových struktur. Periodická okrajová podmínka Periodic boundary condition Pokud bychom chtěli simulovat dynamiku proteinu, je vhodné protein obalit vodou. Aby voda neuletěla někam do vesmíru, byla vymyšlena periodická okrajová podmínka. Studovaná molekula se nachází v kvádru naplněném molekulami vody. Pokud několik takovýchto identických kvádrů naskládáme na sebe, tak na sebe svými plochami navazují jako "molekulární puzzle". Pokud při simulaci nejaká molekula vyjede stěnou do sousedního kvádru, pak se odpovídající molekula vynoří z protilehlé stěny (sousední kvádr je přece identický). Existuje i PBC kde místo kvádru použijeme mnohostěn. 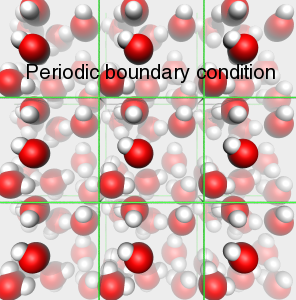 Phylip Phylip Program pro studium fylogenetických vztahů. Podobnost Similarity Pro objasnění pojmů identita, podobnost a homologie, viz Homologie. Poissonova-Bolzmannova rovnice Poisson-Bolzmann equation Obdoba Coulombova zákona, která umožňuje vypočítat elektrostatický potenciál okolí molekulárního systému i v prostředí které není homogenní z hlediska dielektrické konstanty (Poissonova rovnice) a která počítá i s tlumením elektrostatických interakcí elektrolytem (Poissonova-Bollzmannova rovnice). Posičně specifická substituční matice Possition-specific iteration matrix (PSSM) Substituční matice "na míru" skupině proteinů (viz. PSI-BLAST). Přiřazení sekvencí Sequence alignment Porovnání dvou nebo více nukleotidových nebo proteinových sekvencí. Vyjadřuje evoluci daného genu. Dvě aminokyseliny které jsou v proteinovém přiřazení pod sebou by měly být v obdobném místě proteinové struktury. Rozlišujeme binární nebo mnohočetné a globální nebo lokální přiřazení. Přístupový kód Accession number Identifikační označení záznamu (např. sekvence) v databázi. Nemusí to být nutně číslo. Procheck Procheck Program pro posuzování kvality prostorových struktur na základě geometrie. Profil Profile V souvislosti s predikcemi prostorových struktur se jako profil označuje nejaká informace pro jednotlivá aminokyselinová residua proteinu, např. tendence být na povrchu proteinu. PROSA PROSA Program pro posuzování kvality předpovězených prostorových struktur (rozmístění polárních a nepolárních aminokyselin a podobně). PROSITE PROSITE Databáze proteinových motivů (viz). ? Protein-ligand docking Predikce, jestli se testovaný nízkomolekulární ligand na daný protein váže, jak silně se váže a jaká je struktura komplexu. Používá se i při vývoji léčiv ve farmaceutickém průmyslu. ? Protein-protein docking Predikce, jestli se dva proteiny na sebe vážou, jak silně se vážou a jaká je struktura komplexu. PSI-BLAST PSI-BLAST Vylepšená iterativní verze programu BLAST. V prvním kole se provede standardní prohledávání databáze programem BLAST. V dalším kole se na základě výsledků vytvoří pozičně specifická substituční matice ušitá na míru dané rodině genů. Prohledávání databáze pomocí této matice je pak citlivější. Q top QM/MM QM/MM Kombinace kvantové a molekulové mechaniky. Kvantová chemie zpravidla nepotřebuje od nás zadat, jaké atomy jsou jak kovalentně svázané. Na druhou stranu je kvantová chemie zpravidla extrémně počítačově náročná. Pokud chceme studovat enzymovou reakci, je možné aktivní místo popsat kvatově mechanicky a zbytek molekulárně mechanicky. R top Receptor Receptor V souvislosti s protein-ligand dockingem se jako receptor označuje ten protein (i když je to třeba enzym). Renderování Rendering Výpočet fotorealistické grafiky. ? Restraint Umělá síla, která drží nějaké atomy v předem určené posici, vzdálenosti a podobně. Může mít tvar harmonického potenciálu. Používá se například při výpočtu prostorové struktury proteinu na základě experimentálních dat z NMR. Atomy s experimentálně změřenou vzdáleností se při vhodné simulaci udržují v dané vzdálenosti pomocí restrainu. Rozpouštědlu dostupná plocha Solvent acessible surface Plocha získaná tak, že kolem molekuly "obtáčíme" sondu, představující rozpouštědlo. Rozpouštědlu dostupná plocha je pak definovaná tak, kam až se dostane střed sondy. S top Sbalování proteinu Protein folding Též svinování. Proces, při kterém se čerstvě synthetisovaný (nebo uměle denaturovaný) protein dostává do nativní konformace. U některých proteinů může být spojeno s chemickými změnami (cis/trans isomerace před prolinem, tvorba disulfidových můstků atd). Může být usnadňován chaperony, chaperoniny a jinými systémy. Silové pole Force field Soubor publikovaných parametrů pro výpočet potenciální energie (a zároveň sil) jako funkce souřadnic atomů. Rozlišujeme "all atom", které popisují systém souřadnicemi všech atomů, "united atom", které popisují malé skupiny atomů (např. CH2) jako jednu částici a "coarse grained", které popisují větší skupiny atomu (např. postraní řetězec aminokyseliny) jako jednu částici. Silová pole jsou zpravidla vyvinuta pro určitou skupinu molekul (proteiny, nukleové kyseliny, cukry atd.). Řada silových polí byla vyvinuta společně se stejnojmeným simulačním programem (AMBER, GROMOS, CHARMM). Simulace Simulation Pro rozdíl mezi modelováním a simulací viz Modelování. Simulace molekulové dynamiky Molecular dynamics simulation Výpočetní metoda, která se snaží vypočítat časový vývoj souřadnic molekulárního systému, (jinými slovy "tetelení" molekul). Základem je počáteční struktura (čas = 0) a vhodná metoda pro výpočet potenciální energie (většinou silové pole, drsnější povahy používají kvantovou chemii či QM/MM). Vlastní simulace je vlastně řešením pohybových rovnic. 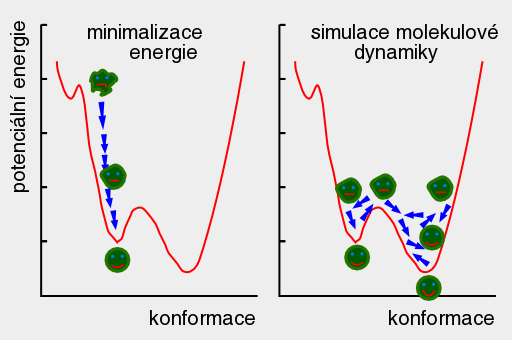 Simulované žíhání Simulated annealing Postup, kdy se při simulaci zvyšuje a snižuje teplota, podobně jako při opravdovém žíhání. Cílem je překonat energetické bariery. Často se simulované žíhání používá jako obecný optimalizační algoritmus.  Skládání Assembly Skládání překrývajících se výstupů ze sekvenátorů do kompaktní sekvence, většinou s využitím vzájemné podobnosti. Používá se u "shotgun" sekvenování. Skóre Score Nějaké číslo, které vyjadřuje např. jak silně se váže ligand na protein, jak kvalitní je model a podobně. Používá se, pokud je tvůrce programu líný tuto informaci vyjádřit fyzikální veličinou (např. změnou Gibbsovy energie vazby ligandu na protein). Substituční matice Substitution matrix Matice, vyjadřující jak jsi jsou podobné jednotlivé aminokyseliny nebo nukleotidy. Používá se při konstrukci přiřazení sekvencí nebo při prohledávání databází se sekvenčním dotazem. Zpravidla byly tyto matice konstruovány porovnáváním sekvencí v nějaké rodině genů. Trochu jinak funguje posičně specifická iterativní matice (PSSM, viz). SwissProt SwissProt Etablovaná moderovaná sekvenční databáze proteinů. Systémová biologie Systems biology Disciplína bioinformatiky, která se snaží získat užitečné informace z hory dat, generovaných genomickými projekty. Nejčastěji se jedná o analýzy sítí interakcí protein-protein a modelování signalizačních a metabolických drah. T top Teplát Template Vzor. Známá prostorová struktura proteinů, která je použita jako strukturní vzor pro předpověď struktury jiného proteinu. Topologie Topology V souvislosti se simulací molekulové dynamiky se pojmem topologie označuje informace o kovalentní struktuře systému, nutná pro konstrukci tvaru rovnice silového pole. Trajektorie Trajectory Záznam simulace molekulové dynamiky. U top UniProt UniProt Jedna z hlavních sekvenčních databází proteinů. Uzel Node Místo v dendrogramu, kde se rozdělují dvě větve. Značí posledního společného předka sekvencí na obou větvích. V top Větev Branch, Clade Spojnice mezi uzly nebo mezi uzlem a vlastní sekvencí. Virtuální screening Virtual screening Postup často používaný při vývoji ligandů, např. léčiv. Vezme se obrovská databáze sloučenin, vytvoří se modely jejich prostorových struktur a pak se tyto molekuly počítačově testují, např. pomocí dockingu. Molekuly, u kterých je předpovězena biologická aktivita jsou pak testovány experimentálně. Při virtuálním screeningu je nutné dělat kompromisy mezi přesností a rychlostí. Tento postup má velký podíl falešně positivních a falešně negativních výsledků, avšak dokáže obohatit databázi o molekuly se skutečnou biologickou aktivitou. Vnějšák Outgroup Sekvence ne moc podobná ostatním, kterou zařadíme mezi sekvence, pro něž počítáme dendrogram. Má za cíl napomoci definovat kořen stromu. Vzorkování Sampling Pokud chceme studovat molekulu, která může existovat v konformaci A a B, pak nás zajímá vzájemná rovnováha. Během simulace molekulové dynamiky by molekula měla zaujímat konformace A a B s pravděpodobnostmi odpovídajícími skutečné rovnováze mezi oběma konformacemi, jinými slovy by měly být možné vypočítat rovnovážnou konstantu z podílu časů, které molekula stráví v konformaci A a B. Problém ale je, že simulační metody jsou počítačově náročné a můžeme simulovat pouze krátké časy. Pokud změna konformace z A do B probíhá v řádu milisekund a my můžeme simulovat pouze nanosekundy, pak máme minimální šancivypočítat rovnovážnou konstantu výše popsaným postupem. O tomto problému se často mluví jako o problému vzorkování. Existují metody, které se snaží tento problém elegantně řešit. |