Molekulární modelování v biochemii
Pod pojmem "molekulární modelování" rozumíme nejen simulace,
ale obecně jakékoliv studie tvaru či konformace molekul. Často
vystačíme "jen" s minimem potenciální energie
(tzv. optimalizace struktury). Typickým úkolem je interpretace
rozptylových a jiných (NMR) experimentů, tzv. refinement, kdy se
struktura stanovená s velkými experimentálními chybami doladí tak, aby
odpovídaly délky vazeb, atomy se nepřekrývaly atd. K tomu všemu
potřebujeme molekulární model neboli silové pole. Výběr modelu je
ovlivněn především snahou maximálně šetřit, protože chceme pracovat
s makromolekulami o velikosti tisíc i více atomů.
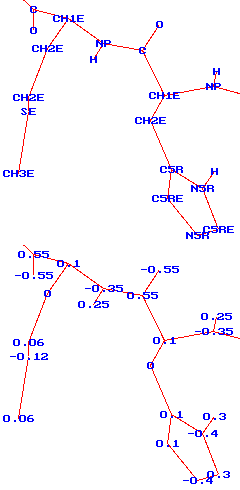 Základním stavebním prvkem modelů je atom či přesněji řečeno atomové
jádro; někdy (zvláště u starších modelů) se alifatické skupiny jako
-CH3 nahrazují jedním centrem ("united" nebo
"extended" atom - viz obrázek vpravo, který představuje
model peptidového řetězce v CHARMM19), výjimečně se naopak přidává
pomocné centrum mimo atomy (model vody TIP4P). Počet typů atomů je
však větší než chemických prvků, protože např. atom uhlíku ve skupině
>C=O má jiné vlastnosti než v benzenu. Atomy také zpravidla nesou
parciální náboje (viz spodní část obrázku).
Základním stavebním prvkem modelů je atom či přesněji řečeno atomové
jádro; někdy (zvláště u starších modelů) se alifatické skupiny jako
-CH3 nahrazují jedním centrem ("united" nebo
"extended" atom - viz obrázek vpravo, který představuje
model peptidového řetězce v CHARMM19), výjimečně se naopak přidává
pomocné centrum mimo atomy (model vody TIP4P). Počet typů atomů je
však větší než chemických prvků, protože např. atom uhlíku ve skupině
>C=O má jiné vlastnosti než v benzenu. Atomy také zpravidla nesou
parciální náboje (viz spodní část obrázku).
Síly mezi atomy se dělí na vazebné (bonded) a nevazebné
(nonbonded). Nevazebné síly působí mezi atomy na různých molekulách a
také mezi atomy jedné molekuly, které nejsou ovlivněny chemickými
vazbami. Atomy oddělené více než třemi vazbami v řetězci
(např. koncové vodíky v propanu
CH3.CH2.CH3) jsou se berou jako
nevázané, atomy spojené vazbou (symbolicky 1-2), dvěma (1-3) či třemi
(1-4) vazbami v řetězci jsou vázané; u posledního případu se kombinují
oba druhy sil.
Nevazebné síly
Nevazebné síly jsou Coulombické a (zpravidla) Lennardovy-Jonesovy.
Vzhledem k šetření se síly pro vzdálenost atom-atom větší než určitá
vzdálenost zanedbávají (viz kap. 5.2.2 skript). Toto je dosti hrubou
aproximací pro elektrostatické síly. Jednou široce používanou
metodou, jak přesnost poměrně levně zlepšit, je nahradit skupiny
(např. >C=O) od určité vzdálenosti bodovým dipólem; silové pole je
totiž obvykle navrženo po skupinách, které jsou elektricky neutrální.
Parametry Lennardova-Jonesova potenciálu se tabelují jako parametry
pro jednotlivé atomy, přičemž křížové parametry (dvojice různých
atomů) se vypočtou z vhodných kombinačních pravidel.
Vazebné síly
Vazby, pokud nejsou v simulaci pevné (viz kap. 7.5.2.), se obvykle
aproximují harmonickým potenciálem
Ubond(r) = (Kbond/2) (r - r0)2
kde r je vzdálenost atomů, r0 její
rovnovážná hodnota a Kbond silová konstanta (tuhost vazby).
Podobně se řeší vazebné úhly, tedy síly 1-3:
Uangle(alpha) = (Kangle/2) (alpha - alpha0)2
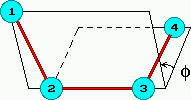 Síly 1-4 jsou dvojí. Vlastní torze (proper torsions) tvaru
Síly 1-4 jsou dvojí. Vlastní torze (proper torsions) tvaru
Udih(phi) = Kdih cos(n phi) ,
kde phi je tzv. dihedrální úhel (viz obrázek vpravo) a n je číslo, např. pro torzní potenciál C-C-C-C v butanu je n=3, případně se sčítá několik členů pro n=0,1,2,3.
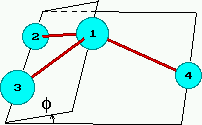 Nevlastní torze (improper torsions) slouží ke "zpevnění"
skupin s sp2 hybridizací jako H2C=O, kde uhlík
leží v rovině s ostatními atomy, případně k zajištění tetraedrické
geometrie tří vazeb okolo skupiny CH, jestliže je tato
reprezentována jedním interakčním centrem (viz CH1E na obrázku nahoře):
Nevlastní torze (improper torsions) slouží ke "zpevnění"
skupin s sp2 hybridizací jako H2C=O, kde uhlík
leží v rovině s ostatními atomy, případně k zajištění tetraedrické
geometrie tří vazeb okolo skupiny CH, jestliže je tato
reprezentována jedním interakčním centrem (viz CH1E na obrázku nahoře):
Uimprop(phi) = (Kimprop/2)
(phi - phi0)2 .
[Zpět na Informace pro studenty, skripta]
[back to J. Kolafa homepage]